NAKAI Lab.
Laboratory of Functional Analysis in silico
Human Genome Center
The Institute of Medical Science
The University of Tokyo







research
(brief summary of selected research activities)
Genome Informatics
ホヤ遺伝子発現調節領域のゲノムワイドな解析で脊椎動物の進化に迫る
Genome-wide identification and characterization of transcription start sites and promoters in the tunicate Ciona intestinalis
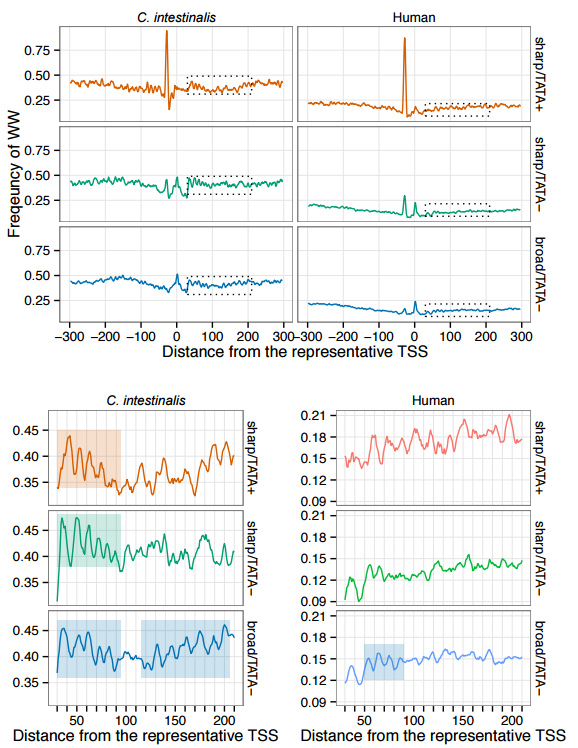 The tunicate Ciona intestinalis has recently emerged as a powerful model organism for gene regulation analysis. Here, using TSS-seq, we identified transcription start sites (TSSs) at the genome-wide scale and characterized promoters in C. intestinalis. Specifically, we identified TSS clusters (TSCs), high-density regions of TSS-seq tags, each of which appears to originate from an identical promoter. TSCs were found not only at known TSSs but also in other regions, suggesting the existence of many unknown transcription units in the genome. We also identified candidate promoters of 79 ribosomal protein (RP) genes, each of which had the major TSS in a polypyrimidine tract and showed a sharp TSS distribution like human RP gene promoters. Ciona RP gene promoters, however, did not appear to have typical TATA boxes unlike human RP gene promoters. Surprisingly, despite the absence of CpG islands, Ciona TATA-less promoters showed low expression specificity like CpG-associated human TATA-less promoters. Using TSS-seq, we also predicted non-operon-type trans-spliced gene TSSs, and found that their downstream regions had higher G+T content than those of non-trans-spliced gene TSSs. This higher G+T content was also observed in downstream regions of operon-type trans-spliced gene TSSs. Although the mechanism of trans-splicing remains unclear, the conservation of high G+T content in two different types of trans-spliced genes may suggest its importance in C. intestinalis trans-splicing.
The tunicate Ciona intestinalis has recently emerged as a powerful model organism for gene regulation analysis. Here, using TSS-seq, we identified transcription start sites (TSSs) at the genome-wide scale and characterized promoters in C. intestinalis. Specifically, we identified TSS clusters (TSCs), high-density regions of TSS-seq tags, each of which appears to originate from an identical promoter. TSCs were found not only at known TSSs but also in other regions, suggesting the existence of many unknown transcription units in the genome. We also identified candidate promoters of 79 ribosomal protein (RP) genes, each of which had the major TSS in a polypyrimidine tract and showed a sharp TSS distribution like human RP gene promoters. Ciona RP gene promoters, however, did not appear to have typical TATA boxes unlike human RP gene promoters. Surprisingly, despite the absence of CpG islands, Ciona TATA-less promoters showed low expression specificity like CpG-associated human TATA-less promoters. Using TSS-seq, we also predicted non-operon-type trans-spliced gene TSSs, and found that their downstream regions had higher G+T content than those of non-trans-spliced gene TSSs. This higher G+T content was also observed in downstream regions of operon-type trans-spliced gene TSSs. Although the mechanism of trans-splicing remains unclear, the conservation of high G+T content in two different types of trans-spliced genes may suggest its importance in C. intestinalis trans-splicing.
Yokomori et al. Genome Res. 26:140-150 (2016)
時系列データを扱うスパース回帰モデルによって免疫機能に関わる介在パスウェイを同定
Discovery of differentially activated intermediary pathways in knockout experiments using sparse regression
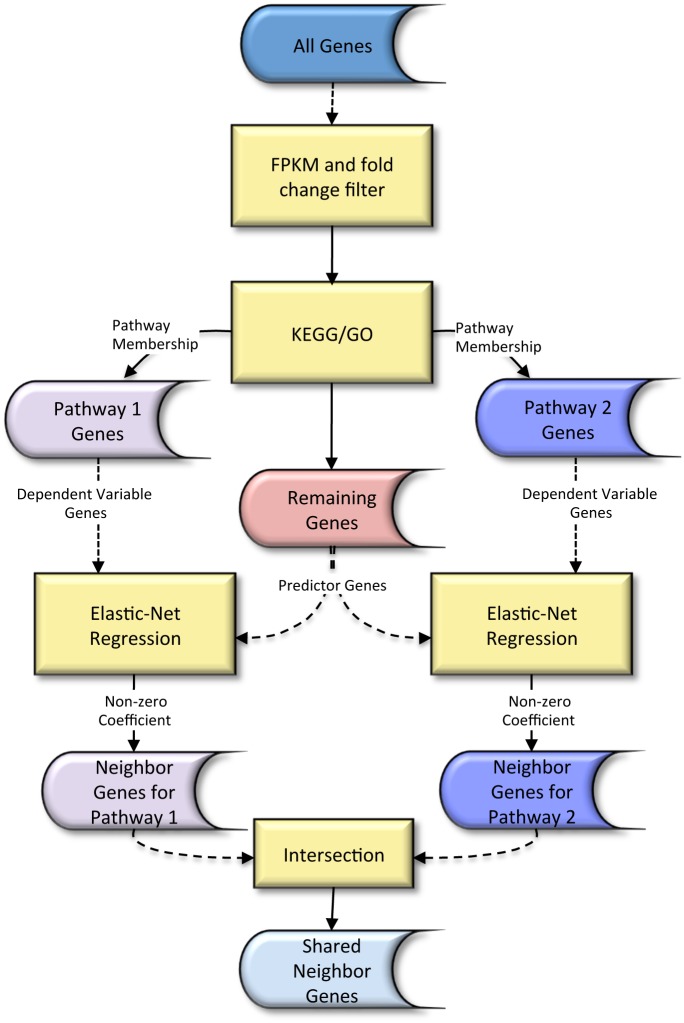 The use of pathways and gene interaction networks for the analysis of differential expression experiments has allowed us to highlight the differences in gene expression profiles between samples in a systems biology perspective. The usefulness and accuracy of pathway analysis critically depend on our understanding of how genes interact with one another. That knowledge is continuously improving due to advances in next generation sequencing technologies and in computational methods. While most approaches treat each of them as independent entities, pathways actually coordinate to perform essential functions in a cell. In this work, we propose a methodology based on a sparse regression approach to find genes that act as intermediary to and interact with two pathways. We model each gene in a pathway using a set of predictor genes, and a connection is formed between the pathway gene and a predictor gene if the sparse regression coefficient corresponding to the predictor gene is non-zero. A predictor gene is a shared neighbor gene of two pathways if it is connected to at least one gene in each pathway. We compare the sparse regression approach to Weighted Correlation Network Analysis and a correlation distance based approach using time-course RNA-Seq data for dendritic cell from wild type, MyD88-knockout, and TRIF-knockout mice, and a set of RNA-Seq data from 60 Caucasian individuals.
The use of pathways and gene interaction networks for the analysis of differential expression experiments has allowed us to highlight the differences in gene expression profiles between samples in a systems biology perspective. The usefulness and accuracy of pathway analysis critically depend on our understanding of how genes interact with one another. That knowledge is continuously improving due to advances in next generation sequencing technologies and in computational methods. While most approaches treat each of them as independent entities, pathways actually coordinate to perform essential functions in a cell. In this work, we propose a methodology based on a sparse regression approach to find genes that act as intermediary to and interact with two pathways. We model each gene in a pathway using a set of predictor genes, and a connection is formed between the pathway gene and a predictor gene if the sparse regression coefficient corresponding to the predictor gene is non-zero. A predictor gene is a shared neighbor gene of two pathways if it is connected to at least one gene in each pathway. We compare the sparse regression approach to Weighted Correlation Network Analysis and a correlation distance based approach using time-course RNA-Seq data for dendritic cell from wild type, MyD88-knockout, and TRIF-knockout mice, and a set of RNA-Seq data from 60 Caucasian individuals.
Liang et al. PLoS One 10:e0137222 (2015)
造血幹細胞における遺伝子発現制御様相を可視化する計算機モデルの構築
Computational promoter modeling identifies the modes of transcriptional regulation in hematopoietic stem cells
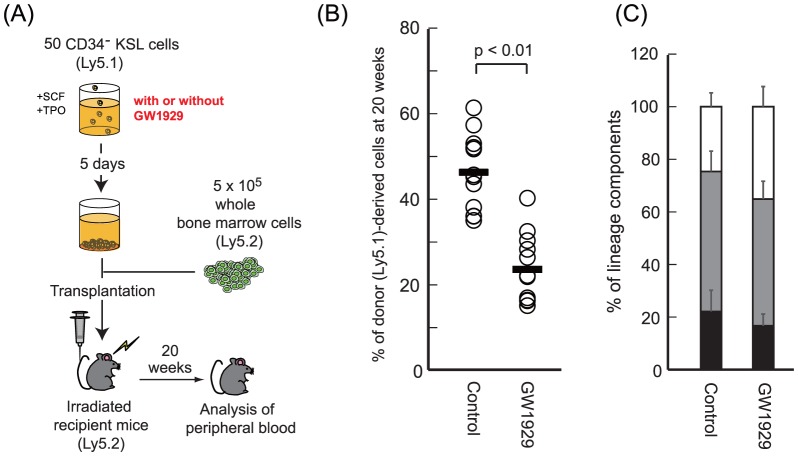 Extrinsic and intrinsic regulators are responsible for the tight control of hematopoietic stem cells (HSCs), which differentiate into all blood cell lineages. To understand the fundamental basis of HSC biology, we focused on differentially expressed genes (DEGs) in long-term and short-term HSCs, which are closely related in terms of cell development but substantially differ in their stem cell capacity. To analyze the transcriptional regulation of the DEGs identified in the novel transcriptome profiles obtained by our RNA-seq analysis, we developed a computational method to model the linear relationship between gene expression and the features of putative regulatory elements. The transcriptional regulation modes characterized here suggest the importance of transcription factors (TFs) that are expressed at steady state or at low levels. Remarkably, we found that 24 differentially expressed TFs targeting 21 putative TF-binding sites contributed significantly to transcriptional regulation. These TFs tended to be modulated by other nondifferentially expressed TFs, suggesting that HSCs can achieve flexible and rapid responses via the control of nondifferentially expressed TFs through a highly complex regulatory network.
Extrinsic and intrinsic regulators are responsible for the tight control of hematopoietic stem cells (HSCs), which differentiate into all blood cell lineages. To understand the fundamental basis of HSC biology, we focused on differentially expressed genes (DEGs) in long-term and short-term HSCs, which are closely related in terms of cell development but substantially differ in their stem cell capacity. To analyze the transcriptional regulation of the DEGs identified in the novel transcriptome profiles obtained by our RNA-seq analysis, we developed a computational method to model the linear relationship between gene expression and the features of putative regulatory elements. The transcriptional regulation modes characterized here suggest the importance of transcription factors (TFs) that are expressed at steady state or at low levels. Remarkably, we found that 24 differentially expressed TFs targeting 21 putative TF-binding sites contributed significantly to transcriptional regulation. These TFs tended to be modulated by other nondifferentially expressed TFs, suggesting that HSCs can achieve flexible and rapid responses via the control of nondifferentially expressed TFs through a highly complex regulatory network.
Park et al. PLoS One 9:e93853 (2014)
自然免疫反応を司る遺伝子ネットワークの時系列変化を推定
Linking transcriptional changes over time in stimulated dendritic cells to identify gene networks activated during the innate immune response
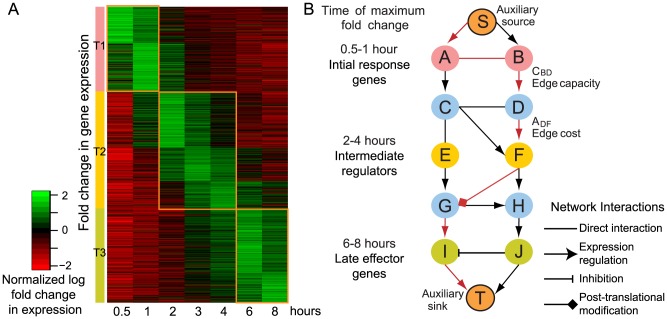 The innate immune response is the first level of protection in organisms against invading pathogens. It consists of a large number of proteins functioning in signaling cascades triggered by the binding of fragments from microbes to specific cellular receptors. Disruptions in these pathways can lead to numerous diseases. As such, the innate immune system has been the subject of a large number of studies. However, due to its complexity and the interplay of a large number of pathways, it is not yet completely understood. In this study, we measured transcriptional changes in activated immune cells and used this information in the context of a large network of protein-protein and protein-DNA interactions to identify a smaller network of response genes. We did this by identifying the most probable network paths connecting genes showing large changes in their expression patterns at successive stages of the response. Analysis of this activated gene network revealed the associations between various temporal regulators of the innate immune response. We also identified response networks for immune cells lacking important mediators, MyD88 and TRIF, to clarify the distinct functional modules affected by their associated pathways in the innate immune response.
The innate immune response is the first level of protection in organisms against invading pathogens. It consists of a large number of proteins functioning in signaling cascades triggered by the binding of fragments from microbes to specific cellular receptors. Disruptions in these pathways can lead to numerous diseases. As such, the innate immune system has been the subject of a large number of studies. However, due to its complexity and the interplay of a large number of pathways, it is not yet completely understood. In this study, we measured transcriptional changes in activated immune cells and used this information in the context of a large network of protein-protein and protein-DNA interactions to identify a smaller network of response genes. We did this by identifying the most probable network paths connecting genes showing large changes in their expression patterns at successive stages of the response. Analysis of this activated gene network revealed the associations between various temporal regulators of the innate immune response. We also identified response networks for immune cells lacking important mediators, MyD88 and TRIF, to clarify the distinct functional modules affected by their associated pathways in the innate immune response.
Patil et al. PLoS Comput. Biol. 9(11):e1003323 (2013)
Computational Proteomics
タンパク質複合体のイン・シリコ機能解析の現状と展望
Methods for protein complex prediction and their contributions towards understanding the organization, function and dynamics of complexes
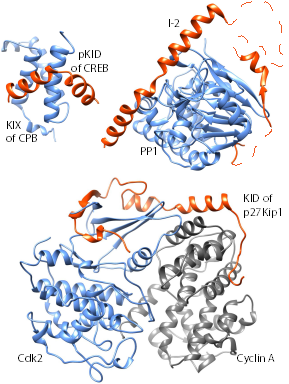 Complexes of physically interacting proteins constitute fundamental functional units responsible for driving biological processes within cells. A faithful reconstruction of the entire set of complexes is therefore essential to understand the functional organisation of cells. In this review, we discuss the key contributions of computational methods developed till date (approximately between 2003 and 2015) for identifying complexes from the network of interacting proteins (PPI network). We evaluate in depth the performance of these methods on PPI datasets from yeast, and highlight their limitations and challenges, in particular at detecting sparse and small or sub-complexes and discerning overlapping complexes. We describe methods for integrating diverse information including expression profiles and 3D structures of proteins with PPI networks to understand the dynamics of complex formation, for instance, of time-based assembly of complex subunits and formation of fuzzy complexes from intrinsically disordered proteins. Finally, we discuss methods for identifying dysfunctional complexes in human diseases, an application that is proving invaluable to understand disease mechanisms and to discover novel therapeutic targets.
Complexes of physically interacting proteins constitute fundamental functional units responsible for driving biological processes within cells. A faithful reconstruction of the entire set of complexes is therefore essential to understand the functional organisation of cells. In this review, we discuss the key contributions of computational methods developed till date (approximately between 2003 and 2015) for identifying complexes from the network of interacting proteins (PPI network). We evaluate in depth the performance of these methods on PPI datasets from yeast, and highlight their limitations and challenges, in particular at detecting sparse and small or sub-complexes and discerning overlapping complexes. We describe methods for integrating diverse information including expression profiles and 3D structures of proteins with PPI networks to understand the dynamics of complex formation, for instance, of time-based assembly of complex subunits and formation of fuzzy complexes from intrinsically disordered proteins. Finally, we discuss methods for identifying dysfunctional complexes in human diseases, an application that is proving invaluable to understand disease mechanisms and to discover novel therapeutic targets.
Srihari et al. FEBS Letters 589:2590-602 (2015)
タンパク質天然変性構造のアミノ酸配列が機能推定に役立つ
Evaluation of sequence features from intrinsically disordered regions for the estimation of protein function
With the exponential increase in the number of sequenced organisms, automated annotation of proteins is becoming increasingly important. Intrinsically disordered regions are known to play a significant role in protein function. Despite their abundance, especially in eukaryotes, they are rarely used to inform function prediction systems. In this study, we extracted seven sequence features in intrinsically disordered regions and developed a scheme to use them to predict Gene Ontology Slim terms associated with proteins. We evaluated the function prediction performance of each feature. Our results indicate that the residue composition based features have the highest precision while bigram probabilities, based on sequence profiles of intrinsically disordered regions obtained from PSIBlast, have the highest recall. Amino acid bigrams and features based on secondary structure show an intermediate level of precision and recall. Almost all features showed a high prediction performance for GO Slim terms related to extracellular matrix, nucleus, RNA and DNA binding. However, feature performance varied significantly for different GO Slim terms emphasizing the need for a unique classifier optimized for the prediction of each functional term. These findings provide a first comprehensive and quantitative evaluation of sequence features in intrinsically disordered regions and will help in the development of a more informative protein function predictor.
Sharma et al. PLoS One 9:e89890 (2014)
天然変性構造のアミノ酸組成から見えるタンパク質機能
Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification
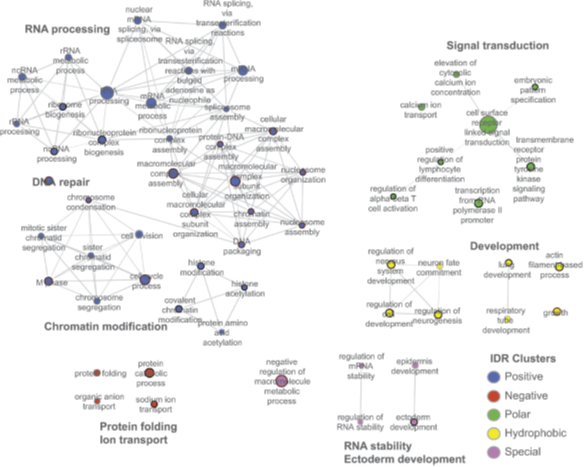 Intrinsically disordered regions in proteins are known to evolve rapidly while maintaining their function. However, given their lack of structure and sequence conservation, the means through which they stay functional is not clear. Poor sequence conservation also hampers the classification of these regions into functional groups. We studied the sequence conservation of a large number of predicted and experimentally determined intrinsically disordered regions from the human proteome in 7 other eukaryotes. We determined the chemical composition of disordered regions by calculating the fraction of positive, negative, polar, hydrophobic and special (Pro, Gly) residues, and studied its maintenance in orthologous proteins. A significant number of disordered regions with low sequence conservation showed considerable similarity in their chemical composition between orthologs. Clustering disordered regions based on their chemical composition resulted in functionally distinct groups. Finally, disordered regions showed location preference within the proteins that was dependent on their chemical composition. We conclude that preserving the overall chemical composition is one of the ways through which intrinsically disordered regions maintain their flexibility and function through evolution. We propose that the chemical composition of disordered regions can be used to classify them into functional groups and, together with conservation and location, may be used to define a general classification scheme.
Intrinsically disordered regions in proteins are known to evolve rapidly while maintaining their function. However, given their lack of structure and sequence conservation, the means through which they stay functional is not clear. Poor sequence conservation also hampers the classification of these regions into functional groups. We studied the sequence conservation of a large number of predicted and experimentally determined intrinsically disordered regions from the human proteome in 7 other eukaryotes. We determined the chemical composition of disordered regions by calculating the fraction of positive, negative, polar, hydrophobic and special (Pro, Gly) residues, and studied its maintenance in orthologous proteins. A significant number of disordered regions with low sequence conservation showed considerable similarity in their chemical composition between orthologs. Clustering disordered regions based on their chemical composition resulted in functionally distinct groups. Finally, disordered regions showed location preference within the proteins that was dependent on their chemical composition. We conclude that preserving the overall chemical composition is one of the ways through which intrinsically disordered regions maintain their flexibility and function through evolution. We propose that the chemical composition of disordered regions can be used to classify them into functional groups and, together with conservation and location, may be used to define a general classification scheme.
Moesa et al. Mol Biosyst. 8:3262-73 (2012)
Bioinformatics Resource Development
大量データの統合処理による高確度のタンパク質相互作用DBを構築
HitPredict: A database of high confidence protein-protein interactions
 The study of genes and their inter-relationships using methods such as network and pathway analysis requires high quality protein-protein interaction information. Extracting reliable interactions from most of the existing databases is challenging because they either contain only a subset of the available interactions, or a mixture of physical, genetic and predicted interactions. Automated integration of interactions is further complicated by varying levels of accuracy of database content and lack of adherence to standard formats. To address these issues, the latest version of HitPredict provides a manually curated dataset of 398 696 physical associations between 70 808 proteins from 105 species. Manual confirmation was used to resolve all issues encountered during data integration. For improved reliability assessment, this version combines a new score derived from the experimental information of the interactions with the original score based on the features of the interacting proteins. HitPredict provides a web interface to search proteins and visualize their interactions, and the data can be downloaded for offline analysis. Data usability has been enhanced by mapping protein identifiers across multiple reference databases.
The study of genes and their inter-relationships using methods such as network and pathway analysis requires high quality protein-protein interaction information. Extracting reliable interactions from most of the existing databases is challenging because they either contain only a subset of the available interactions, or a mixture of physical, genetic and predicted interactions. Automated integration of interactions is further complicated by varying levels of accuracy of database content and lack of adherence to standard formats. To address these issues, the latest version of HitPredict provides a manually curated dataset of 398 696 physical associations between 70 808 proteins from 105 species. Manual confirmation was used to resolve all issues encountered during data integration. For improved reliability assessment, this version combines a new score derived from the experimental information of the interactions with the original score based on the features of the interacting proteins. HitPredict provides a web interface to search proteins and visualize their interactions, and the data can be downloaded for offline analysis. Data usability has been enhanced by mapping protein identifiers across multiple reference databases.
López et al. DATABASE bav117 (2015)
マルチオミックス情報基盤としての転写開始点データベースDBTSS
DBTSS: Database of Transcriptional Start Sites
Suzuki et al. Nucleic Acids Res. 43:D87-91 (2015)
バーチャル顕微鏡で多能性幹細胞由来のテラトーマを観察・共有する
OpenTein: a database of digital whole-slide images of stem cell-derived teratomas
 Human stem cells are promising sources for regenerative therapy. To ensure safety of future therapeutic applications, the differentiation potency of stem cells has to be tested and be widely opened to the public. The potency is generally assessed by teratoma formation comprising differentiated cells from all three germ layers, and the teratomas can be inspected through high-quality digital images. The teratoma assay, however, lacks consistency in transplantation protocols and even in interpretation, which needs community-based efforts for improving the assay quality. OpenTein has been designed as a searchable, zoomable and annotatable web-based repository system. We have deposited 468 images of teratomas derived by our transplantation of human stem cells, and users can freely access and process such digital teratoma images.
Human stem cells are promising sources for regenerative therapy. To ensure safety of future therapeutic applications, the differentiation potency of stem cells has to be tested and be widely opened to the public. The potency is generally assessed by teratoma formation comprising differentiated cells from all three germ layers, and the teratomas can be inspected through high-quality digital images. The teratoma assay, however, lacks consistency in transplantation protocols and even in interpretation, which needs community-based efforts for improving the assay quality. OpenTein has been designed as a searchable, zoomable and annotatable web-based repository system. We have deposited 468 images of teratomas derived by our transplantation of human stem cells, and users can freely access and process such digital teratoma images.